- Project summary. 1.Project Title
Chemically Programmed Immunity and Adaptive Vaccinology
This One Health proposal is based on an invention by the Nobel Laureate Dr Kary Mullis (December 28, 1944-August 7, 2019). The platform consists of nucleic acids (RNA, DNA, or single-stranded or double- stranded of either or both) chemically modified to provide a linker through a nanoparticle or nanofiber with nuclease resistance; the multivalent aptamers bind pre-existing immune cell receptors or natural or induced antibodies through the nanostructures to other aptamers pre-selected in vitro to bind to a moiety on the pathogen of interest. The pre-existing, anti-ligand activity is intended to re-direct an effective immediate immune response (including pre-existing successful vaccination technology) against the pathogen targeted by the in vitro selected nucleic acid binding sequence. This linker construct is in some instances intended to trigger further immune memory for future antibody, cell immune, toll-like receptor, or other adaptive and innate immune responses through a natural cascade, providing for autogenous vaccine development. The second part of this proposed platform consists of a vector delivery system consisting of the linker synthetic nanoparticles or nanofibers and the synthetic nucleic acid aptamers linked in such a way to retain the above activity upon release of the aptamer that will facilitate dermal, oral, nasal, respiratory, or parenteral delivery of the effective nucleic acid. In the best example of this complete platform, the composite will transform or transfect a target cell for the mass production of a biosynthetic counterpart of the fully synthetic composite, which performs the same anti-pathogen, anti-parasite function, allowing for scale-up by fermentation for manufacture or in vivo amplification. Parasite pathogens have been selected for the targets under this proposal because of the difficulty and underserved need for developing vaccines against them and because animal models, and in some cases side-by-side in field zoonotic disease, allow for animal field testing under conditions which are most appropriate for predicting a human response from animal data. This composite structure also allows for tracer technology to be incorporated while maintaining function in order to determine the biological fate of such composites in vivo and in situ.
2. Platform Description and Rationale:
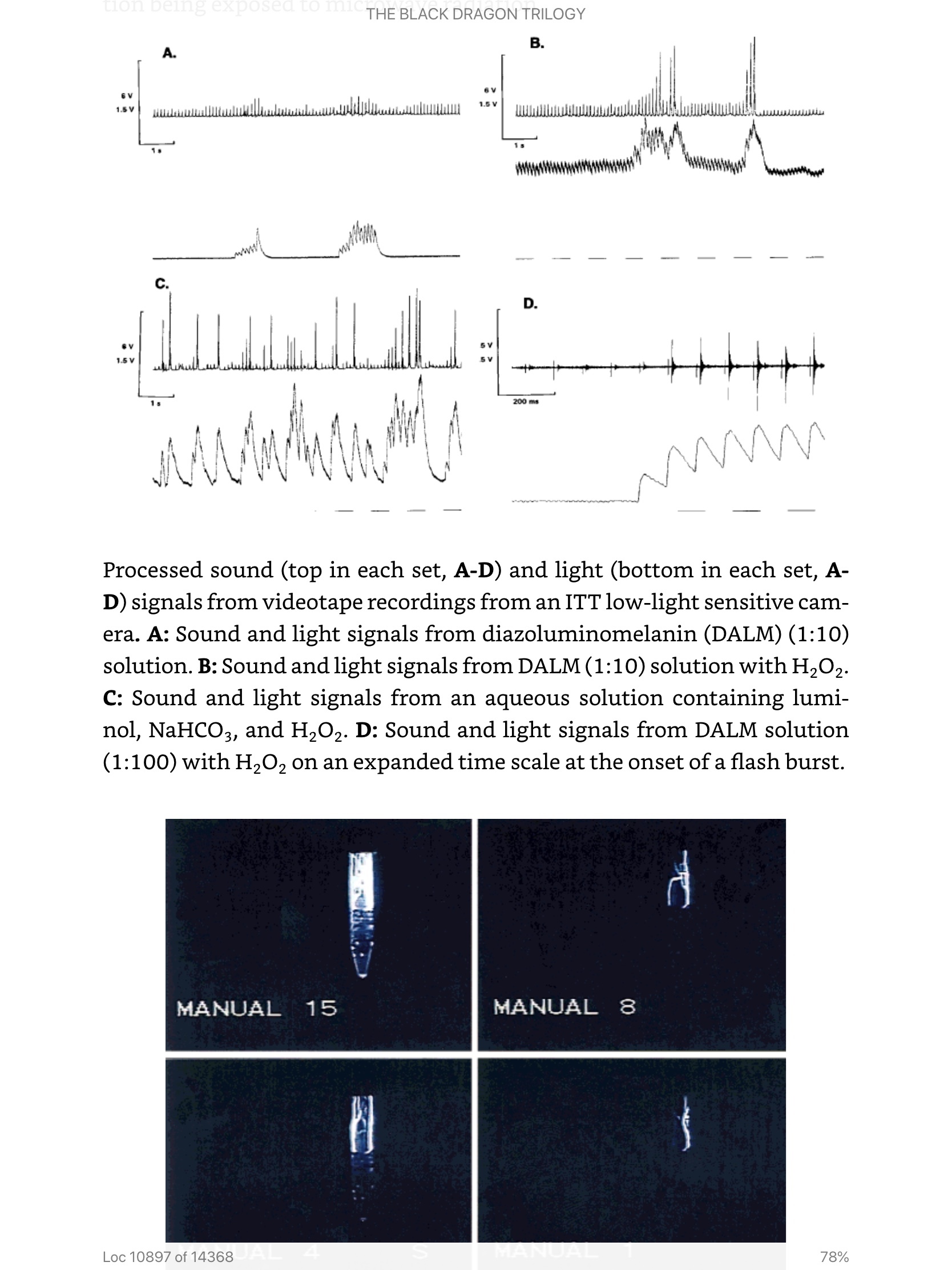
The platform has been described in detail in enabling US and international patents and peer reviewed scientific papers. The platform and examples will be briefly described here. The details will be given by reference to save space in this expression of interest. The technology is presented in two complementary parts: (1) the nucleic acid aptamer with a chemically conjugated ligand to recruit an existing immune response to natural or vaccine-derived immunogen attached to an aptamer which links the sequences previously selected for DNA binding in vitro to the target pathogen or parasite; and (2) the synthetic nanoparticle/nanofiber vector to carry the aptamers into the host and target tissues by a convenient port of entry, releasing them at the nidus of infection or infestation. Also, a selection process and ruggedized prototype device for use under field conditions to collect target pathogens and isolate an appropriate binding aptamer for amplification and therapeutic use has been designed and undergone preliminary tests. A nanosprayer for topical and respiratory/mucous membrane delivery has also been developed; however, several types of nanospray delivery systems are now commercially available (for cosmetics). The same aptamers and vectors designed for therapeutics have been initially designed for diagnostics using fluorescence dequenching, thermochemiluminescence, magnetic capture, electron microscopy contrast media, and visible light microscopy staining and marking and diagnostically demonstrated against spotted fever type rickettsia, Bacillus anthracis, botulinum toxin, Francisella tularensis (in the field to confirm collection and isolation), and Shiga toxin. Furthermore, the synthetic vectors have been made which transfect and transform bacterial, animal and human cells, traceable by PCR, gene expression, fluorescence, and staining. The bacterial hosts have shown not only the physical and biological evidence of genetic transfer, but also subsequent production of transferable genetic material and vector biosynthetic polymer and transfer of the genetic material to naïve bacterial hosts and their transfer in the same way for three iterations. This biosynthetic form was also transferable to human and animal cells and detected by PCR. The transfer polymer made synthetically or biosynthetically was diazoluminomelanin, a poly (hydroxy) ortho phenylene, made from 3-amino-tyrosine or tyrosine by nitration, reduction and diazotization. This polymer when activated absorbs a wide range of electromagnetic radiation from UV to visible light to microwave and radiofrequency radiation. The polymer has a high affinity for DNA, protects it from nucleases and radiation damage until saturated (high energy pulsed radiation) then can cut or destructively damage DNA or RNA by free radical generation. The microwave killing mechanism has been demonstrated against anthrax and other bacilli spores. Biosynthetic polymer containing the transferable genetic material is readily transferred to bacteria, animal and human cells.
Phenotypic consequences of nanoparticle composite transformation or transfection and fail-safe system for its destruction have been demonstrated.
Data for treatment of anthrax in mice using Sterne and Ames strain Bacillus anthracis has been collected. The data for immediate recruitment of the immune system in mice were very robust because although survival of the more pathogenic strain was limited, one must keep in mind that the anthrax was delivered by inhalation at very high multiple lethal doses and the aptamer was delivered most effectively after a 3 hour delay by inhalation and was only against the protective antigen of the anthrax toxin and had to mobilize the immune system within 3 days with total lethality at 4 days. Alpha 1,3 galactose aptamer was the immune linker, but the mice do not make antibody against this epitope and human serum, containing natural anti-gal antibody had to be used to direct the mouse immune system after immunization of the mice with human serum. Independent experiments at the US Army Medical Institute of Infection Disease showed that lung clearance of spores was superior in the presence of aptamer: approximately 67 CFU (colony forming units) of bacilli per gram of lung of a treated mouse vs an estimated 44,600 CFU bacilli per gram of lung of an untreated mouse, a 665-benefit ratio.
No human in vivo toxicity data are available, only in vitro data with human cells. However, experiments were also performed using the FETAX (Frog Embryo Teratogenesis Assay: Xenopus) procedure, which showed DALM to be non-toxic. Clinical trials with polymeric nanoparticles and DNA vaccines which are very similar to these agents have been performed with favourable results and some are still underway (see references). DNA vaccines have been shown to be well tolerated and safe.
3. Research and Technical Objectives – Statement of work
Objectives
The objectives of this proposal are to demonstrate the effectiveness of the platform technology and delivery systems described in this proposal in appropriate in vitro and in vivo models, safety and efficacy of such a demonstrably effective form against a clinically significant parasitic disease with both natural animal and human correlates, first in clinical animal models then in human clinically derived samples, and finally in human subjects.
4. Endpoints
The endpoints are to produce a highly reproducible and clinically relevant, for safety and efficacy, composite nanoparticle aptamer platform which can be readily re-directed toward many infectious and parasitic targets by mere substitution of components without extensive re-testing and field trials.
5. Target pathogens
Pathogen 1: Malaria (Plasmodium falciparum and others): There is no current effective vaccine for this disease and resistance to the most effective treatment with artemisinin is spreading. However, natural resistance has been demonstrated in endemic areas in children, but the effective response requires IgM and not IgG and the target antigen galactose-alpha-1,3-galactose. Vaccines that lead to IgG response development will not be as effective as those that direct the immune system toward an IgM response to alpha gal.
Pathogen 2: Heartworms (Dirofilaria immitis): this disease is not only pervasive in canines across the world, but the current treatment uses the same macrocyclic lactones, to which resistance is developing and spreading, used in human filariasis; macrocyclic lactones are the only class of drugs currently available to treat these filarial diseases, which means the development of resistance would eliminate any effective treatment, and they involve altering or blocking the release of an antigen which interferes with the immune response to these filaria and which is effectively activated upon treatment to eliminate microfilaria. There is a robust antibody response which is interfered with by soluble antigen/antibody complex formation which could be overcome by the appropriate redirection or facilitation of immune targeting.
Pathogen 3: Dracunculus medinensis, Guinea worm; the hope to eradicate this parasite, now only in Africa, has recently shown signs of failure after finding that the once thought to be rare infection in dogs (over a thousand infected dogs discovered) is now commonly present in them in Chad. This infection of dogs in areas of re-emerging human infection allows for studies of the platform response in dogs under field conditions which would be most relevant to concurrent human disease.
6. Pre-Clinical Approach:
The worst case non-clinical/pre-clinical development approach has already been established by previous Department of Defence research on anthrax spore protection using a mouse/Sterne/Ames anthrax spore inhalation and immediate mobilization of the immune system by inhalation aptamer composites and looking for prolonged survival and clearing of inhaled anthrax spores. The dose ranges have been pre-determined by in vitro neutralization molar ratio data (for anthrax toxins but of course not the proposed targets). The general toxicity issues are the activation of toll-like receptor innate immunity and direction toward Th1 vs Th2 immune pathways (i.e., avoiding alpha gal meat allergy-like responses by appropriate cellular immune cell targeting along with parasite targeting) which can be adjusted by making alterations in the nucleic acid sequences of the aptamers. Since the immune response can be recruited during concomitant infections, the reduction in microfilaria or other circulating parasitic forms (i.e., in malaria), we can directly assess the response in an individual human or animal as its own control. - 7. Clinical development plan and regulatory strategy:
The strategy for clinical development through Phase I of the proposed pathogens uses constructs which are essentially variations of the same form and these lead to certain derived, in-depth analyses of immunological mechanisms of action of the platform and toxicity. Each pathogen/parasite addressed will take a similar approach by substitution: the anti-malarial aptamer composite will contain a bifunctional nanoparticle coated with a nucleic acid with galactose-alpha-1,3-galactose moiety at the exposed end to engage natural human antibody against the alpha gal and an anti-B1 lymphocyte receptor(s) aptamer, and/or including anti-IgM aptamers, on the same nanoparticle surface to directly stimulate IgM production for the removal of the malarial parasites. These composites can be tested against peripheral human monocytic/lymphocytic cells first (binding and stimulation assays) and antibody in human serum for binding. The Phase I test would look at the toxicity of the alpha gal/anti-B1 lymphocyte aptamer nanoparticle composite. Initially, a model form will be tested (for toxicity and efficacy) in the avian model of malaria infection by using poultry immunized against alpha gal and a nanoparticle with an alpha gal nucleic acid moiety and an anti-avian IgY aptamer. The poultry will be infected with avian malaria and the various component controls and complete platform. The anti-heartworm platform, which can be extended to human filarial parasites by substitution of appropriate aptamer specific parasite or general cross-reacting filarial antigen moieties, will consist of a bifunctional nanoparticle coated with an aptamer selected against the endosymbiont Wolbachia and another aptamer against one or more excretory/secretory antigens (currently used as diagnostic antigens in dogs). Domestic (pet) dogs with and without concurrent heartworm infections from endemic heartworm areas will be recruited for treatment/vaccination with the preparation following in laboratory testing of efficacy and toxicity in dogs and monitored for microfilaria and immunological responses indicating adult female heartworm presence. The anti-dracunculid composite will be composed of the same nanoparticle types as tested above, species specific anti-excretory/secretory antigens aptamer(s) for the parasite (many have already been cloned) and an anti-measles/distemper virus or anti-measles/distemper antibody epitope aptamer. Cross reacting anti-measles/distemper aptamers will allow the same composite prep to be used in both dogs and humans, who have been immunized against these viruses and will either be challenged with the currently available vaccines at time of treatment or those demonstrating high antibody titers against these viruses at the time. The planned licensure pathway for approval of a phase 1 clinical trial will be through a San Antonio clinical trial firm to be hired. - 8. Mechanism of action:
Because the mechanism involves re-directing an immune response from an ineffective but pre-existing abundant one, the reduction in numbers of immature circulating forms of the parasites or adult parasite fertility and viability will be used to assess the effectiveness of the mechanism of action. Furthermore, levels of isotypes and idiotypes of antibodies and specific committed cellular immunity will be assessed by quantity and specificity of response to the effective antigens/immunogens. The reappearance of disease in an endemic area in individuals (animals or humans) treated and cleared in this way, by adaptive vaccinology, will be assessed for reappearance of infection over time and the extent of signs and symptomology of apparent disease. The effectiveness of re-treatment will be evaluated and the necessity to further re-direct the response. - 9. Chemistry, Manufacturing & Control (CMC) Development
The large-scale manufacture of the platform involves methods already being used in commercial production of aptamers (SELEX) and newer methods described in the publications and patents referenced, including an automated method, and a field diagnostic system for finding binding sequences and cloning them for fermentation production. - 10. Timeline and Key milestones:
Timeline – Decision/key milestones
From initiation of project: 3-6 months to select aptamers for potential molecular targets; starting after selection/conjugation of aptamers to nanoparticles and testing of binding activity, determining quality and quantity reliability (first milestone that must be passed): 6 months; testing in in vitro cellular models, animal and human, and laboratory models if necessary (mice and chickens or other poultry for malarial responses; dogs for microfilaria and adult heartworm responses; dracunculid responses will have to be done in the field in Chad in dogs); at least one, and preferably, the first two laboratory animal models must be passed without overt toxicity to proceed. By 2.5 years from the initiation, at least phase 1 trials should begin for one of the specific anti-parasite composites. Phase 2 and 3 will most likely exceed the 3-year limit of this funding proposal. - 11. Vaccine Development Timeline: Because changing the target for vaccination only requires a substitution into the platform, human trials could occur within 3 months of identifying a target. The approval of prior tests of the DNA/nucleic acid and nanoparticle platforms for use in humans as safe and the concurrent use of similar preparations in domestic animals (under USDA/APHIS biologicals rules) or in-country animals would greatly accelerate the process.
The aptamer platform composite approach does not require the prior development of immunity against the target organism but employs pre-existing or concomitant immunity to provide protection. In anthrax trials with naïve mice at multiple lethal doses of anthrax spores, immune protection against lethal anthrax toxin was initiated as early as 2 hours after exposure and maintained in an advancing infection at periods of 24 hours until 14 days when primary immunity took over. This worst case would not be expected with the parasites described since they are not uniformly fatal at 4 days as the overwhelming anthrax spore challenge would be. Also, the parasites themselves, when present as initial larval forms or subsequent adults maintain an active but ineffectual immune response, which the aptamer composite platform would re-direct for the elimination of the parasites. The presence of the nanoparticle-based aptamer composite platform is anticipated to continue for at least a year and it remains to be seen if the re-directed immune pathway will persist after the composite platform has broken down, but this is possible if not probable and will be explored as part of the experimental phase with subsequent re-challenges with parasites. Potentially no boosting will be required. - 12. Scale up to target doses
The estimated time of release, if anti-ligand targets are already available for aptamer binding selection, and if no clinical trials are required before such an emergency response (except for safety), then the best time would be 3 months and worst 6 months, starting from the beginning of the process. If the aptamer sequence is already known, production and delivery could occur in 2-4 weeks. The platform is designed for single dose use at best, but can be used, as it was with anthrax, in multiple serial doses for severe overwhelming potential lethal disease. The questions about manufacture process have been addressed elsewhere in this pre-proposal. - 13. Budget
Total budget estimate
The initial pre-clinical development approximate cost will be at least $1.8M per year, which includes all the manufacturing technology development for scale, initial in vitro cell culture and animal testing. It will also include testing for toxicity of the nanoparticle and nucleic acid constructs unless data can be accepted on similar DNA and nanoparticle constructs already being used in human and animal clinical trials corresponding to this purpose (if not this may raise the cost to $3M yearly). The production of the scaled-up end-product for each parasite for use in testing, clinical trials and/or emergency response would be at least $550,000 for the custom made (not final manufacturing cost) for 100,000 doses. The cost objective for the final product for distribution for medical and veterinary use is 16 to 25 cents a dose at today’s costs of intermediate scale up manufacturing of nucleic acids (of bifunctional target length aptamers) and nanoparticles, or as close to this cost as possible. The major pass through costs would be for clinical trials, if they are to be funded all or in part, about $55M. Otherwise work up to that time (over a minimum of 3 years) would be $5.9M to $6.55M.
Additional funding not applicable. - 14. Application organization/consortium
Experience and track record of applicant organization and cooperating partner(s)
John G. Bruno: B.S., Microbiology with Chemistry Minor, Univ. of Arizona (1985) , Ph.D., Microbiology with emphasis on Immunology, Univ. of Arizona, Tucson (1991), Jan 2003-August 2018 Senior Scientist, Vice President and Chief Science Officer- Operational Technologies Corporation (OpTech), OTC Biotechnologies, LLC, and Pronucleotein Biotechnologies, Inc., San Antonio, TX., September 2018 – Present, Principal Scientist and Director of Biotechnology – Nanohmics, Inc., Austin, TX., 68 Grants/Contracts; Funding > $16M; 104 Peer-Reviewed Journal Articles; 16 Total U.S. and WIPO/PCT/EU Patents .
Ronald M. Cook: UC Berkeley College of Chemistry (BS 1969); University of Washington, Seattle, Department of Chemistry, Ph.D. 1974; postdoctoral, University of California, San Francisco, (Dept of Microbiology and Immunology), 1974-76; founded Biosearch, Inc in 1977 to supply synthetic peptides (enkephalins) and protein bioconjugates for research; 1980, synthesis of nucleic acids, Biosearch became leading innovator in synthetic DNA oligonucleotide chemistry; 1982, the company developed first commercially viable Oligo synthesizer (SAM I); one of the first SAM units was placed with Kary Mullis, at Cetus, enabling the invention of PCR in 1983; 1990-1993, Principal Consultant to Beckman Instruments; 1993- 2015, Biosearch Technologies, Inc.: President/CEO, Chief Technical Officer; Chairman, Light Speed Genomics, a Santa Clara company; Chairman, DNA Technology, a Danish oligo-manufacturing company; Chairman, Vitra Bio, a German manufacturer of specialty glass for solid phase chemistry; consultant and/or served on Scientific Advisory Boards of Beckman Instruments, Solexa (Cambridge, England, RMC was a co-founder), developing next generation DNA sequencing technology; and Mosaic (Cambridge, Ma), developing solid phase PCR and contingent diagnostic applications; 2015-2019. LGC-Genomics, Chief Scientific Officer, Chairman, Co-Founder Optical Biosystems.
George W. Irving III; DVM (1965), MS (1970), Diplomate American College of Laboratory Animal Medicine (1972), and American College of Veterinary Preventive Medicine (1977); served 30 years as USAF Military Officer, and 23 Years as VP at Conceptual MindWorks, Inc., directing research in Chemical & Bio Warfare Agent defense, Non-Lethal Weapons Bio Effects, and Pharmaceutical/Vaccine Development.
Johnathan L. Kiel: DVM (1974, Texas A&M); PhD (Microbiology and Biochemistry, 1981, Texas Tech); Diplomate American College of Veterinary Microbiologists (1984), Charter Diplomate in Veterinary Parasitology (2011); served 38 years total, as a military officer, as a Civil Servant (GS14-15) research veterinarian, and Senior Scientist (Brigadier General Equivalent) in the Senior Executive Service in electromagnetic bioeffects and biosurveillance and counterproliferation of biological warfare and other infectious agents; 106 peer-reviewed articles; 30 patents. The fundamental form of this technology was invented by Dr Kary Mullis (deceased 7 August 2019), the recipient of the Nobel Prize, for inventing, PCR in 1993; with his collaboration, the platform was extended to other applications and delivery systems by the researchers of the United States Air Force at the Air Force Research Laboratory. - 15. Dissemination plan of study results and data sharing:
Peer-reviewed journal papers will be used when applicable to share data. Clinical trial registration and studies will be managed by a San Antonio clinical trials firm (such as Clinical Trials of Texas, Inc.). Because the targets for this project involve tropical diseases as well, we will reach out to public health and medical authorities of potential target countries such as Chad and other African countries through the World Health Organization and the OIE, World Organisation for Animal Health. All patient data will be protected in accord with US and international laws. Patent applications, both US and international, will be used to disclose the processes, but non-exclusive licensing will be used as much as possible to make the technology globally available. Procedures in accord with the USDA/APHIS will be followed to license the resulting animal biologicals produced from the project platform. - 16. References.
- 1. Mullis, K.B., Vivekananda. J., Kiel, J.L., Cook, R.M. Chemically Programmable Immunity, US Patent 8,604,184 B2, December 10, 2013.
- 2. Kiel, J.L., Tijerina, A., Holwitt, E.A., Sloan, M.A., Woitaske, M., and Fan, M. Compositions, Methods and Uses for Biosynthetic Plasmid Integrated Capture Elements, US Patent 8,628,955 B2, Jan. 14, 2014.
- 3. Kiel, Johnathan L., Holwitt, Eric A., Fan, Michael (Maomian), Roper, Shelly D., Methods and Compositions for Rapid Selection and Production of Nucleic Acid Aptamers, US Patent 9,273,345 B2, March 1, 2016.
- 4.Vivekananda, J., and Kiel, J. L. Anti-Francisella tularensis DNA aptamers detect tularemia antigen from different subspecies by Aptamer-Linked Immobilized Sorbent Assay, Laboratory Investigation 86: 610-618, 2006.
- 5. Fan, M., McBurnett, S. R., Andrews, C. J., Allman, A. M., Bruno, J. G., and Kiel, J. L. Aptamer Selection Express: A Novel Method for Rapid Single-Step Selection and Sensing of Aptamers. J Biomol Tech 19(5), 311–319 (December 2008).
- 6. Bruno, J G. Aptamer–biotin–streptavidin–C1q complexes can trigger the classical complement pathway to kill cancer cells In Vitro Cell.Dev.Biol.—Animal (2010) 46:107–113.
- 7. Mutapi, F., Billingsley, P.F., and Secor, W.E. Infection and treatment immunizations for successful parasite vaccines. Trends in Parasitology March 2013, Vol. 29, No. 3, 135-141.
- 8. Bruno, J.B., Richarte, A.M., Savage, A.A., and Sivils, J.C. Development and Characterization of DNA Aptamers Which Bind Kinesins from Leishmania Promastigotes. J. Bionanosci. 8, 1–12, 2014.
- 9. Aguilar, R., et al. Antibody responses to α-Gal in African children vary with age and site and are associated with malaria protection. Scientific Reports. 3 July 2018, DOI:10.1038/s41598-018-28325-w.
- 10. Kristian, S.A., Hwang, J.H., Hall, B., Leire, E., Iacomini, J., Old, R., Galili, U., Roberts, C., Mullis, K.B., Westby, M., and Nizet, V. Retargeting pre-existing human antibodies to a bacterial pathogen with an alpha-Gal conjugated aptamer. J Mol Med (Berl). 2015 June; 93(6): 619–631.
- 11.Slatko, B.E.,Taylor, M.J., and Foster, J.M. The Wolbachia endosymbiont as an anti-filarial nematode target. Symbiosis (2010) 51:55–65.
- 12. Pati, R., Shevtsov, M., and Sonawane, A. Nanoparticle Vaccines Against Infectious Diseases. Frontiers in Immunology (9), October 2018, Article 2224.
- Don’t lose sight of the versatility of this approach (just because the original proposal involves its use against parasites),especially for COVID-19, where the path the immune response takes is the difference between recovery or death or long term chronic disease.
Author: Dr. Johnathan Kiel
Settling a Paradox for COVID-19: Cytokine Storm or Not?
The new definition of sepsis is as life-threatening organ dysfunction caused by a dysregulated host response to infection more closely fits severe COVID-19. It abandons use of host inflammatory response syndrome criteria (SIRS) in identification of sepsis and eliminated the term severe sepsis. It is usually associated with bacterial rather than viral infections, but this new definition fits severe COVID better than cytokine storm. This latter description suggests a simple positive correlation and causation with pro-inflammatory cytokines. One should expect as their systemic levels go up, the symptoms get worse. The measurable level should correlate with the severity. The alternative is, if this doesn’t happen, the severity must be related to direct viral effects and immunosuppression, allowing the virus to greatly increase in number and spread to many organs, reeking havoc. At first glance these mechanisms may appear diametrically opposed. However, measurements can reflect the dynamics of cytokine levels verses progression of symptoms and exhaustion of the immune system not being synchronized. A recent study illustrates this paradox. https://www.thelancet.com/action/showPdf?pii=S2213-2600%2820%2930404-5. In spite of clinically resembling other classic syndromes associated with cytokine storms, meta-analysis of COVID-19 studies, published or posted preprints, between Nov 1, 2019, and April 14, 2020, in which interleukin-6 concentrations in patients with severe or critical disease were determined (25 COVID-19 studies of 1245 patients were included) showed lower than expected cytokine levels measured. The study groups included four each in sepsis (n=5320), cytokine release syndrome (n=72), and acute respiratory distress syndrome unrelated to COVID-19 (n=2767). Cytokine release syndrome may occur after treatment with some types of immunotherapy, such as monoclonal antibodies and CAR-T cells. Cytokine release syndrome is caused by a large, rapid release of cytokines into the blood from immune cells affected by the immunotherapy. I had seen this with intraperitoneally injected immobilized oxidase-peroxidase causing the rapid experimental destruction of large Novikoff abdominal hepatocarcinoma cancers in rats. In patients with severe or critical COVID-19, the pooled mean serum interleukin-6 concentration was 36.7 pg/ml. Mean interleukin-6 concentrations were nearly 100 times higher in patients with cytokine release syndrome (3110.5 pg/mL), 27 times higher in patients with sepsis (983.6 pg/mL), and 12 times higher in patients with acute respiratory distress syndrome unrelated to COVID-19 (460 pg/ml). Other cytokines gave similar levels with a mean TNFα (tumor necrosis factor alpha) concentration of 5.0 pg/mL (2.3–10.7 pg/mL) in patients with COVID-19, mean concentration of 34.6 pg/mL (20.0–59.9 pg/mL) in patients with sepsis and 52.2 pg/mL (2.0–1390 pg/mL) in patients with cytokine release syndrome. All but one COVID-19 study of the studies analyzed had mean TNFα concentrations lower than 10 pg/mL. IFNγ (gamma interferon) concentrations were not elevated in patients with COVID-19, at an average of 10.8 pg/mL, but were highly elevated in patients with cytokine release syndrome, averaging 3722.1 pg/mL (624–21838 pg/ml). Mean sIL-2R (serum soluble interleukin 2 receptor) was elevated in patients with COVID-19, but much less than in patients with cytokine release syndrome comparison (506 pg/mL vs 12396 pg/ml). The disparity between studies in TNF alpha level reported in an earlier post suggests that when it is measured in the course of the disease is important, but where it is measured is also important. Innate immunity is often very localized, and therefore, not measurable accurately in the serum. CRP (C reactive protein) concentrations were similar in COVID-19 and sepsis patients, and higher in patients with cytokine release syndrome. The studies examined indicated that patients with COVID-19 had higher D-dimer elevations (local fibrin formation and lysis are part of the inflammatory response, and fibrin degradation products, including D-dimer, have been shown to have diverse effects on inflammatory processes and acute phase responses, including hepatic synthesis of acute-phase proteins, such as CRP and fibrinogen) than did patients with sepsis. Mean ferritin and lactate dehydrogenase (measures of cell damage) concentrations were much higher in patients with cytokine release syndrome than in those with COVID-19, but still highly elevated in patients with COVID-19. Again, this indicates that pro-inflammatory processes were in play somewhere other than reflected in the blood measurements. In other highly lethal infectious disease which should resemble sepsis and septic shock syndrome, surprises have been found in cytokine stochastic measurements https://www.ncbi.nlm.nih.gov/pmc/articles/PMC303127/. Edema toxin (ET) of anthrax bacteria and lipopolysaccharide of Gram negative bacteria (LPS) each induced human monocytes to secrete approximately the same amounts of IL-6. ET did not inhibit and, in most experiments, modestly enhanced LPS-induced IL-6 production. In contrast to this stimulation of IL-6 production, ET yielded little or no TNF-alpha production, which is associated with septic shock. Moreover, ET profoundly inhibited LPS-induced TNF-alpha synthesis. Monocytes treated with dibutyryl cAMP, an active analog agonist of cAMP, produced cytokines in a pattern identical to that of cells treated with ET. The disruption of cytokine networks as a consequence of unregulated, ET-mediated cAMP accumulation in human monocytes may impair cellular antimicrobial responses and contribute to clinical signs and symptoms of anthrax. Lethal toxin of anthrax proteolytically inhibits MAP kinase kinase, an essential enzyme in the activation pathways of lymphocytes (MAPKK1 and MAPKK2 are cleaved which inactivates MAPKK1 and inhibits the MAPK signal transduction pathway). Another study with anthrax, that I was involved in, examined these two toxins that cause an intense systemic inflammatory response, edema, shock, and eventually death and showed similar dysfunction and dysregulation of the immune system not expected based on the effects on cytokine function. https://europepmc.org/article/med/11890668. Parent strain mice and knockouts were challenged with LD95 of anthrax spores (5 x 10exp6). administered subcutaneously. The results showed that all genetic knockouts succumbed to anthrax infection as frequently as the parent. TNF antibody delayed death but TNF receptor 1 knockout genetic modification had no effect. IL-1 receptor or iNOS (inducible nitric oxide synthase) knockouts died sooner. Anthrax was more abundant in the injection site of TNF-alpha and iNOS knockouts compared to parent suggesting that localized attenuated inflammatory cellular response increased the disease progression. The deaths were so rapid that, with the exception of edema and necrosis at the injection sites, no pathological changes were observed in internal organs, indicating a lethal per acute shock syndrome. Therefore, the dysregulation leading to dysfunction of the immune system in severe COVID-19 is not unheard of. The general distribution of an otherwise localized response may be the real culprit in severe COVID-19. As mentioned in a previous post, when anti-parasitic immune responses are suddenly generalized, shock and death can rapidly and irreversibly ensue. This is seen with crushing of parasitic fly larvae in a wound (myiasis) or rapid disintegration of filarial or other parasitic worms in a host.
To summarize. the possible sequence of events in COVID-19 are to first infect nasal and oral epithelial cells, trigger interferon and mucosal immunity; if this does not clear the virus, it descends into the lungs, widely infecting pneumocytes, leading to low grade pneumonitis that may trigger an effective adaptive response or not. If the adaptive response fails or results in autoantibody to interferons, then viremia spreads the virus systemically and causes lymphocytopenia (decrease in lymphocyte counts in blood, with removal of lymphocyte regulation of the immune response, typical of viremia). Multi-organ infection and generalized infection of the lungs, followed by immune-mediated cellular (antibody-dependent) inflammatory response against all the infected sites leading to sudden, generalized collateral damage that would otherwise be local, but because of the widespread multiple sites leads to severe disease and even multiple organ failure and death.
New Method Supports the Possibility that Fluorescent Aptamer Nanoparticles could Yield Potentially the Fastest Handheld COVID-19 Diagnostic Test
A rapid diagnostic method for SARS-CoV-2, which provides results in about 5 minutes, based on DNA oligonucleitides labeled with fluorescent dyes binding to viruses, which, in turn, undergo microscopic image analysis for identification has been reported https://www.medrxiv.org/content/10.1101/2020.10.13.20212035v2.full.pdf. Aptamers are synthetic oligonucleotides selected for specifically binding to infectious agents or many other targets. They have been labeled with fluorescent dyes for tracing the binding and further developed into double-stranded DNA probes, where a complementary DNA strand has a fluorescent quencher that when displaced allows the fluorescence to be observed https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2628068/pdf/JBT-05-311.pdf . Therefore, the Oxford method could be enhanced by the rapid aptamer method.

No “Magic” in “New”: Immune Response to SARS-CoV-2 is More Characteristic than First Thought: More Encouraging Data
It appears protective immunity to SARS-CoV-2 may last at least 6 months and up to more than a year. Although the strain changes recently seen might be the reason for the rare re-infections recently reported, I believe it is more. The evidence for protective cross immunity with other CoVs and even bacterial antigens (pneumonia vaccines) rules (see earlier post) against the small genetic changes (D614G in the spike protein) in the spike protein inhibiting immunity. New data indicates that the mutation actually increases antibody neutralization https://www.medrxiv.org/content/10.1101/2020.07.22.20159905v2.full.pdf. I suspect that it is because the first viral infection was cleared early by interferon 1, IgA, and/or IgM, the latter which decline within 3 months. It is the IgG, particularly IgG3, that lasts longer: 7 months and the memory B cells even longer. But if infection response clears virus in less than 2 weeks, the IgG response never happens—opening up for reinfection. Researchers at Massachusetts General Hospital found IgG levels remained elevated for four months in those who recovered from COVID-19, and were protective neutralizing antibodies, which decreased over time. The researchers also showed that those infected with SARS-CoV-2 had immunoglobulin A (IgA) and immunoglobulin M (IgM) responses that were relatively short-lived, declining within about two and a half months or less, on average. https://immunology.sciencemag.org/content/immunology/5/52/eabe0367.full.pdf. Researchers at the University of Toronto discovered IgG antibodies were detectable for up to 115 days (approximately 4 months) in those recovered from COVID-19 https://immunology.sciencemag.org/content/immunology/5/52/eabe5511.full.pdf. University of Arizona focused on antibodies that bind to two different parts of the SARS-CoV-2 virus, most antibody tests look for antibodies at S1, which includes the receptor-binding domain by which the spike protein binds to a protein receptor to infect cells. But they also looked for antibody to the S2 region of the spike protein. They found SARS-CoV-2 antibodies in blood for at least 5 to 7 months, although they believe immunity actually lasts longer. They knew that people who were infected with SARS coronavirus-1, which is the closest relative to SARS-CoV-2, still show immunity 17 years after infection. If SARS-CoV-2 is anything like the first one, they expect antibodies to last at least two years, and it would be unlikely for anything less https://www.cell.com/immunity/pdf/S1074-7613(20)30445-3.pdf?_returnURL=https%3A%2F%2Flinkinghub.elsevier.com%2Fretrieve%2Fpii%2FS1074761320304453%3Fshowall%3Dtrue.
Evidence that Supports Re-directing the Immune System with Aptamers (Chemically Programmable Immunity, Dr Kary Mullis) would Work Against COVID-19: “Until a coronavirus vaccine is ready, pneumonia vaccines may reduce deaths from COVID-19”
https://news.yahoo.com/until-coronavirus-vaccine-ready-pneumonia-191844263.htmlThe higher the rate of pneumococcal vaccination, the lower COVID-19 morbidity and mortality. Vaccination rates with Bacillus Calmette–Guerin, poliovirus, and other vaccines do not affect these COVID-19 numbers, nor do COVID-19 case or death rates follow the numbers of people with diabetes, obesity, or who are over 65. Infant protection may be due to maternal antibodies and antiviral proteins in milk such as lactoferrin that are known to protect against coronavirus infections. Protection might be further provided by rates of Haemophilus influenzae type B (Hib) (universal in infants) and pneumococcal vaccination, the latter varying widely among nations for infants, at-risk adults, and elderly.
The huge differences in the incidence of severe COVID-19 cases and deaths from one nation to another might be due to population density, number of elderly, how and when social distancing restrictions or other protective measures are implemented. It is difficult to explain differences in COVID-19 death rates that vary from between 650 per million to 65 per million in neighboring European nations or death rates as low as 0.5 per million in some Asian nations. The rate of vaccination against Haemophilus influenzae type B and pneumococcal infections by Streptococcus pneumoniae may make the difference in preventing fatal COVID infection outcomes. https:onlinelibrary.wiley.com/doi/epdf/10.1002/bies.202000076. One last caveat, association does not prove causation. This beneficial effect could be extended or enhanced by using aptamer linkers with the current vaccine antigen at one end and the aptamer, selected in vitro for binding to the SARS-CoV-2 (spike) antigens, at the other end of the aptamer, Chemically Programmable Immunity.
Persistence of SARS-CoV-2 on Surfaces, Fomites, All Depends on Conditions and Initial Amount
A recent carefully done laboratory experiment has been published https://virologyj.biomedcentral.com/articles/10.1186/s12985-020-01418-7 . The information needs to be put in context to assess real field environmental survival of SARS-CoV-2. Viruses, in some sense, survive better environmentally than vegetative bacteria but less well than bacterial spores. The rule of thumb is the “dirtier the virus, the longer it survives inactivation”. The cleaner it is, the less it survives. However, as stated in my earlier post, the initial amount can lead to even longer or shorter apparent survival due to exponential decay and the threshold of the detection methods: viral culture in vitro or true infectious dose in a susceptible host. The paper to be discussed used the in vitro method to assess virus viability and vigorous extraction methods (worst case transmission) not touch transfer or re-aerosolization (which are more hazardous to do in the lab). They also used a standard biological matrix (mimic of human expelled biological material) and maximum inoculum dose (based on assumed human expelled dose) which is appropriate to produce optimal survival data in the laboratory. Real life conditions are messier and UV light (all light) was excluded from the experimental conditions. They observed half lives of between 1.7 and 2.7 days at 20 °C, decreasing to a few hours when temperature was elevated to 40 °C. Using initial viral loads approximately equivalent to the highest titers excreted by infectious patients and in a simulated biological matrix, viable virus was isolated for up to 28 days at 20 °C from common surfaces such as glass, stainless steel and both paper and polymer banknotes. In contrast, infectious virus survived less than 24 h at 40 °C (104 F) on some surfaces. Fomite transmission is highly efficient for transmission (efficiencies of 33%) for both fomite to hand and fingertip to mouth transfer for bacteria and phages (no data for CoV). The inoculum was 10 microliters of the suspension (final concentration of 3.38 × 105/10 µL). A single relative humidity set point (50%) was maintained for experiments at 20 °C, 30 °C, 40 °C, respectively, although dew point would have been a more accurate measure since relative humidity changes with temperature. Three replicates of each surface type were inoculated and sampled at the following time points; 1 h, 1 day, 3 days, 7 days, 14 days, 21 days and 28 days post inoculation. For the 40 °C experiment, triplicate samples were inoculated for the following time points; 1 h, 1 day, 2 days, 3 days, 4 days, and 7 days. For non-porous surfaces, for each replicate, virus was eluted in 2 × 115 µL volumes of DMEM (cell culture medium) with repeated pipetting then titrated individually, in quadruplicate wells on a 96-well plate. The recovery from porous surfaces, such as cotton cloth, which are likely to inhibit transfer and recovery under field conditions, was accomplished by soaking swatches of the cloth in 500 µL DMEM and pipetting repeatedly for at least 1 min before 230 µL of the recovered eluent from each swatch was titrated separately, in quadruplicate. Suspensions of Vero E6 cells (3 × 105/mL) were added to the wells and the plates were incubated for 3 days at 37 °C with 5% CO2 to look for cell damage (cytopathic effect) from viral replication in the cells. The majority of virus reduction on cotton occurred very soon after application of virus, suggesting an immediate adsorption effect. The calculated 90%, 1 log, inactivation of virus for surfaces at 20 °C (room temperature = 25 degrees C) ranged from 5.5 days for cotton to 9.1 days for paper notes. At 30 °C, infectious virus was recoverable for 7 days from stainless steel, polymer notes and glass, and 3 days for vinyl and cotton cloth. For paper notes, infectious virus was detected for 21 days, although there was less than 1 log of virus recovered for both 14 day and 21 day time points. At 40 °C, virus recovery was reduced compared to both 20 °C and 30 °C experiments. Infectious SARS-CoV-2 was not recovered past 24 h for cotton cloth and 48 h for all remaining surfaces tested. Greater than 4-log reduction (99.99% reduction from starting titer) was observed in less than 24 h at 40 °C on all surfaces. All calculations were based on linear decay fits, not exponential decay. Again, as stated in an earlier post, the latter model is what is observed under field conditions for other biological agents. Also, CoV is less durable than other viruses ( non-enveloped) such as adenovirus, picornaviruses (can persist for up to a year in the environment) and Norovirus (diverse group of single-stranded positive-sense RNA, non-enveloped viruses belonging to the family Caliciviridae). What needs to be kept in mind here is that incident surface contamination does not equal transferable host absorbed infectious dose. Smooth non-porous surfaces are more likely to transfer virus than porous surfaces that adsorb and bind virus. This is borne out by the low transmission rate (approximately 10% infected) for most exposed populations. However, the paper indicates that we should err on the side of caution and rigorously disinfect potentially contaminated surfaces.
Why “Reinfection” Happens with SARS-CoV-2, or Does It? Answer: Yes (in certain cases)
I discussed in earlier posts that immunity can be incomplete, even with recovery; inappropriate, leading to severe disease; long lasting with cellular mediated immunity; or side-stepped or even made to make things worse when infected by a different strain of the same virus. Also, viruses and other microbes can hide in sequestered privileged sites in the body to re-emerge to spread to other sites before the immune system kicks in again or be shed and spread to other susceptible hosts. Herpes viruses such as those that cause cold sores, genital lesions and chicken pox, followed years later by shingles, hide in nerve endings, only to re-emerge when adaptive immunity declines. Bacterial diseases like brucellosis recur producing undulant fever over years by hiding in cells where immunity or antibiotics have a hard time getting. Leptospira hide in the kidneys and shed in the urine to infect humans and other animals, giving them active leptospirosis, after the shedding host has recovered from acute disease and is generally immune but not in the kidneys. Syphilis moves through 4 phases as immunity to this microbe develops: acute genital form; secondary systemic rash form (skin rashes and/or mucous membrane lesions: sores in the mouth, vagina, or anus; symptoms of secondary syphilis may include fever, swollen lymph glands, sore throat, patchy hair loss, headaches, weight loss, muscle aches, and fatigue); latent (hidden) stage of syphilis, a period of time when there are no visible signs or symptoms of syphilis; and the tertiary fatal central nervous system form (can appear 10–30 years after infection was first acquired; affects multiple organ systems, including the brain, nerves, eyes, heart, blood vessels, liver, bones, and joints). Each step it becomes more and more hidden from the immune system or exhausts the immune responses in the final stage. Perhaps, albeit to a much lesser extent, CoV uses some of these strategies to maintain infections in or among individuals. The change in strain internally could also occur if there is sufficient immune selection pressure to accelerate mutation of the virus (see earlier post on monoclonal antibody treatments). No matter which above mechanism has come to play, re-infection with or re-activation of SARS-CoV-2 has occurred https://www.medrxiv.org/content/10.1101/2020.09.22.20192443v1.full.pdf. In Brazil, two cases of “re-infection” has occurred with two different variants, with increased severity, in one case, associated with increased viral load https://www.preprints.org/manuscript/202101.0132/v2. One additional note, as public health fails and more and more virus is present and carelessness allows larger viral doses to be transmitted, these mechanisms, no matter how rare, become more likely to occur and are amplified.
Good News in Face of Bad: The Immune System is Smarter than We Think: Anticipation, Response and Memory of SARS-CoV-2
In recent studies of COVID patients, of the inflammatory mediators analyzed in serum, IL-6, IL-10, monocyte chemoattractant protein-1 (MCP-1), and interferon-gamma induced protein 10 (IP-10) were the ones significantly increased and correlated with disease severity. No significant changes in other cytokines or chemokines measured at the time of hospitalization with symptoms, including IFN-gamma, IL-1-beta, IL-8, or tumor necrosis factor-alpha (TNF alpha) were seen in these patients. High neutrophilia (numbers of blood neutrophilic granulocytes) appeared in severe COVID-19, because of their role in acute respiratory distress syndrome. Elevated C Reactive Protein (CRP) and IL-6 in patients, when entering hospital, later resulted in death, and increased IP-10 was seen in those who later developed severe disease. IP-10 is an interferon- inducible chemokine that aids directed migration of many immune cells. https://immunology.sciencemag.org/content/immunology/5/51/eabd6197.full.pdf.
Buying time until adaptive immunity develops appears to be where things go wrong in COVID-19. In very lethal diseases, like inhalation anthrax, it progresses too rapidly, 2-4 days, to lethality, for any adaptive immunity to develop, even though vaccination against anthrax is very effective but the initial vaccination response takes about 2 weeks and the anamnestic (booster) response requires 3 days. This latter is just enough to stop a lethal response if the dose of spores inhaled is not too high. If it is, it requires antibiotics as well as prior vaccination over as long as a couple of months. Although anthrax is far more lethal than COVID-19, it appears if the innate immunity (interferon and perhaps monocytic responses) fail too quickly before adaptive cellular and appropriate antibodies kick in, the disease becomes severe and even lethal. As was learned from treating secondary bacterial infections with HIV, antibiotics don’t cure anything without an immune system, they only buy time, or until they fail from bacteria developing antibiotic resistance. The similar failure with COVID-19, is a result of both an inadequate innate immunity and an inappropriate adaptive immunity which accelerate the previous failure by making autoantibodies against interferons, which otherwise block viral replication. The initial dose of virus and the failure to inhibit its replication with antivirals can lead to the same end. IL-6, IP-10, and MCP-1 levels were generally the highest in patients around the time of entering the hospital but decreased rapidly if they proceeded to intensive care, which probably indicates exhaustion of innate immunity.
The good news is that if adaptive immunity is given the time to appropriately develop, it becomes enduring. Preinfection and postinfection patients developed IgG and IgG memory B cells (MBCs) reactive to SARS-CoV-2 proteins. Most importantly, the studies demonstrated that infection generates both IgG and IgG MBCs against the SARS-CoV-2 unique receptor binding domain and the conserved S2 subunit of the SARS-CoV-2 spike protein, shared by other human coronaviruses. This means even after antibody levels decline or become undetectable, long-lived MBCs remain to bring about rapid anamnestic antibody production. The study results also suggested that SARS-CoV-2 infection strengthens preexisting broad immunity to coronavirus, in general, through S2-binding antibody and MBC formation. https://mbio.asm.org/content/mbio/11/5/e01991-20.full.pdf.
Peeking Behind the Curtain: New Insight into How the Immune System Makes a Choice in COVID-19
Recent evidence has arisen, giving at least some insight, into how the immune system can reduce COVID-19 into a mild, unimpactful infection and then, in some cases, progress into a high mortality and morbidity form. Some hint was given by its early history of causing a mild phase progressing into a severe phase after 10-12 days of symptomatic infection, if including 2 days or more of asymptomatic infection, about the length of time (14 days or more) to develop a full humoral response. The most compelling case is for the development of humoral autoimmunity, autoantibody. Patients with life-threatening COVID-19 pneumonia have neutralizing IgG autoantibodies against Interferon-ω, the 13 types of interferon-α, or both, at the beginning of severe disease; a few also have autoantibodies against the other three type I Interferons. The autoantibodies neutralize the ability of the type I interferons to block SARS-CoV-2 infection in cell cultures. These autoantibodies are not found in those with asymptomatic or mild SARS-CoV-2 infection and only rarely in healthy individuals https://science.sciencemag.org/content/sci/early/2020/09/23/science.abd4585.full.pdf. This mechanism also explains the Kawasaki-like syndrome, the multisystem inflammatory syndrome (MIS-C), seen on rare occasion in young children and adolescents. The development of effective and lasting immunity appears to depend on cellular-mediated immunity (CMI; largely, T lymphocytes and production of gamma interferon) https://www.biorxiv.org/content/10.1101/2020.05.26.115832v1.full.pdf. The serum concentrations of IL-17A (a protein that recruits monocytes and neutrophils to an inflammatory site) and IFN-γ, but not TNF-α or IL-6, decrease with age of patients; therefore, gamma interferon’s ability to prevent viral replication but not induce oxidative damage to host cells, as with TNF-α, is more important in children’s responses. Recent evidence supports decline in soluble IL17A receptors, which block TNF-alpha and interferon gamma production by competing with the cellular T17 lymphocyte receptor, leads to more severe COVID19 https://science.sciencemag.org/content/sci/early/2020/10/14/science.abe9403.full.pdf. Adults show a more robust T cell response to the viral spike protein compared to pediatric patients shown by increased expression of CD25+ on CD4+ T cells and the frequency of IFN-γ+CD4+ T cells. Also, serum neutralizing antibody titers and antibody-dependent cellular phagocytosis are higher in adults than in pediatric COVID-19 patients. The neutralizing antibody titer increases with age while the IL-17A and IFN-γ serum concentrations decrease. Therefore, neutralizing antibody levels alone are not sufficient to decrease severity of COVID. The first effective response to the SARS-CoV-2 virus is innate, involving the triggering of interferon 1 production (a complex of 13 proteins) leading to predominantly interferon omega, a form that recruits upregulation of the phagocytic activities of whole blood cells, macrophages and NK (Natural Killer) cell activities with decreased viral excretion https://www.karger.com/Article/Fulltext/495897#top. Type 1 interferons induce type III interferon (IFN-λ), which is primarily restricted to mucosal surfaces and is thought to confer antiviral protection without driving damaging proinflammatory responses. The interferon 1 released from infected cells prevents neighboring cells from replicating virus. The phagocytes produce NO, nitric oxide, that yields inhibition of viral replication and in greater amounts cell death, which may result in killing cells before virus can replicate, and damaging viral RNA. Greater amounts of NO can lead to swelling and further non-specific inflammation if it gets out of control. In children and those adults who recover from mild infection, the virus is eliminated by these innate and adaptive cellular immunity. If virus persists and disseminates in the lungs and other tissues, a strong humoral adaptive response may ensue. If with autoantibodies, phagocytes, with accompanying cytokine inflammation, will be directed against infected tissues, causing sepsis and perhaps death https://www.the-scientist.com/news-opinion/the-immune-hallmarks-of-severe-covid-19-67937. Antibody directed enhancement (ADE) may also ensue, wherein antibody increases viral uptake by white blood cells and endothelial cells by linking the viral bound antibody to the target cells through Fc receptors or complement component linkers/anti- ligands on the cells, and leading to more viral replication https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7165974/pdf/IMR-268-340.pdf.
There may be correlates in animals. Cats get an enteric coronavirus which has little or no signs, but, in certain young cats, it becomes disseminated, causing peritonitis with lethality usually 100%. CMI provides an effective immune response that resolves the infection in the enteric, intestinal form. If it does not, a strong humoral response kicks in and selects for a more pathogenic form of the virus which infects and replicates in macrophages https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7112361/pdf/main.pdf. Also, autoantibodies form. There seems to be genetic predisposition toward this resulting in peritonitis https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2819880/. Peritonitis pathology involves severe systemic inflammatory damage of serosal membranes and widespread pyogranulomatous lesions in the lungs, liver, lymph tissue, and brain. The host immune system produces profound T-cell depletion from the peripheral and lymphatic tissues and changes cytokine expression. The clinical finding of hypergamma-globulinemia-associated feline infectious peritonitis is indicative of over production of antibody against an antigen which cannot be cleared, like a host cell-associated antigen. Therefore, like COVID-19, the feline coronavirus virus disease has this dichotomy, based on viral dissemination and autoantibody. The selection of the appropriate immune pathway with vaccination becomes very important to avoid adverse effects. Other viruses have had a similar warning due to overt responses, predominantly in children, resulting in hemorrhagic disease, when becoming immune (for life) to one strain but then becoming infected with another as in Dengue (4 strains), a mosquito-borne virus. The right antigen for the right vaccine must be selected to prevent such adverse effects. Another way that the viral replication and subsequent damage can be reduced is to stop or delay an overt response without altering it directly by using the anticoagulant heparin which blocks virus’ binding to the target cells https://www.cell.com/action/showPdf?pii=S0092-8674%2820%2931230-7. Stopping selecting the self-destructive immune pathway is what Dr Mullis’ chemically programmed immunity, mentioned in an earlier posting, sought to accomplish as well.
Loss of Wild Places: Ultimate Cause of Infectious Diseases like COVID-19
We are motivated by our emotions, sometimes logic and unfortunately, many times, by our economic desire to accumulate objects, wealth, to guarantee our and our family’s security and future. We do not see that the resources of the Earth are finite, and that continued unlimited growth is unsustainable. To promulgate this illusion for some means others (humans, animals, and plants) must surrender resources to the point they suffer harm, often irreparable and for generations.
But does this belief in our privilege as a superior species really relieve our consciences or relieve us from the responsibility for or consequences of global domination and exploitation? Wildlife populations have plunged by 68% between 1970 and 2016, and only 25% of the planet can still be considered ‘wilderness’ according to the World Wildlife Fund and the Zoological Society of London. There are 8.7 million multicellular species on Earth, 6.5 million on land and 2.2 million in the oceans. Broken down into multicellular groups:
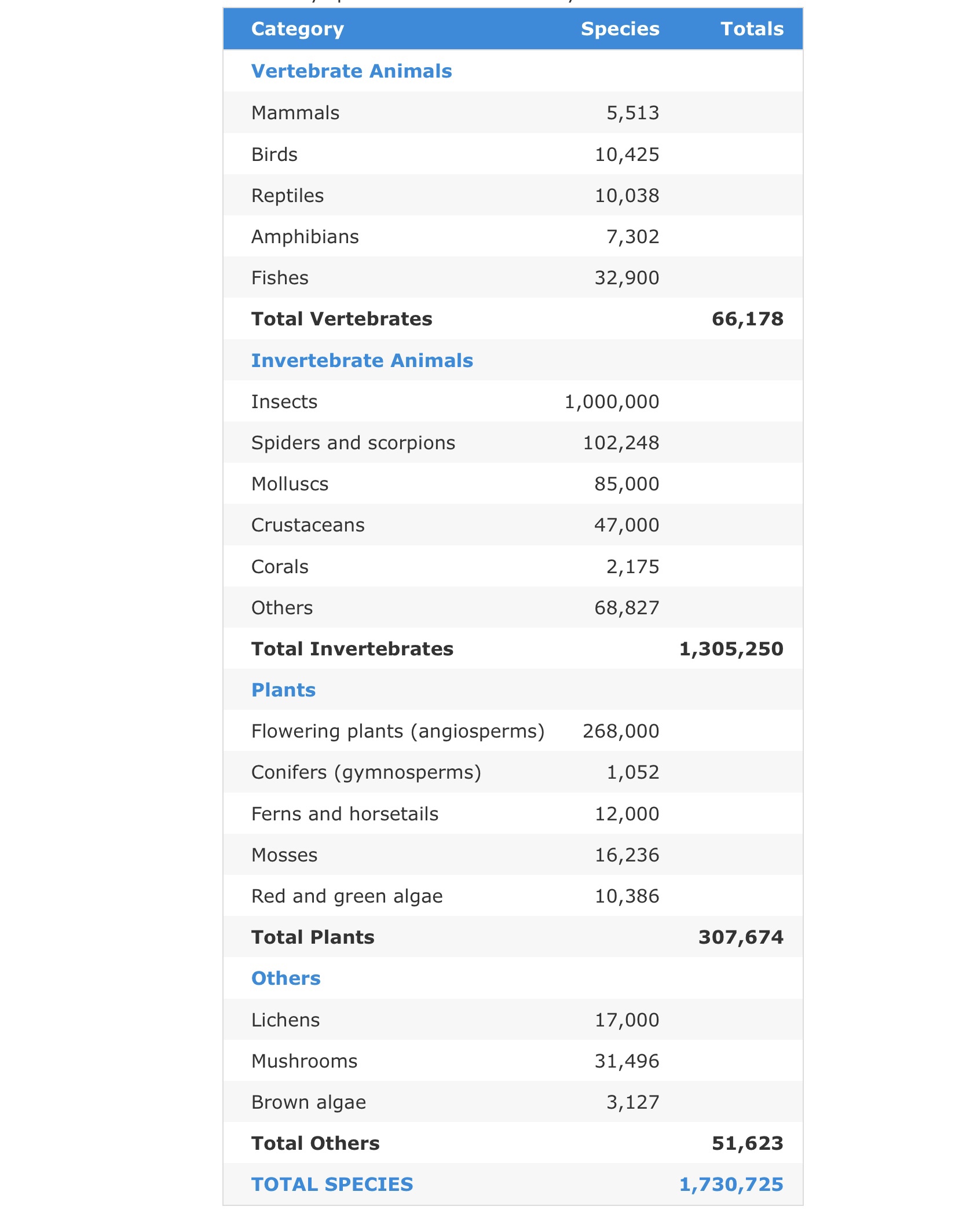
Reference
The World Conservation Union. 2014. IUCN Red List of Threatened Species 2014.3. Summary Statistics for Globally Threatened Species. Table 1: Numbers of threatened species by major groups of organisms (1996-2014).
Our current nemesis, SARS-CoV-2, and its brethren viruses, which may or may not be considered living things, are in numbers that are multiples of the above for every species, even bacteria, Archea, and some giant viruses, which have them. To represent all species on Earth, we would need a “Noah’s Ark” representing approximately 2 × 10(exp)12 tonnes (1 tonne = 1 metric ton = 1000 kg) of biomass of approximately 5 × 10(exp)30 living cells.
All these species are in danger, including human beings. We are entering the 6th mass extinction, this time caused by humans. This would not be the first time humans have faced near extinction.
“It began greater than 160,000 to 135,000 years ago from South Africa and then spread up along the east coast of Africa to the Horn of Africa and then both east and west. The westerly route took it to the Congo River Basin where it spread to the West Coast of Africa and to the talapoin monkeys, the most probable origin of modern VPV (Viper Plague Virus) through rock python predation and tick spread to the smaller ball pythons of West Africa and then to the Vipers co-mingled with them and their tick vectors in the pet trade shipments to Florida (where the modern descendant of this virus was discovered in the early 2000’s). The Easterly route to North and South America took much longer and almost wiped out the human population traveling east. They followed the mid-eastern route to the coast of India to Southeast Asia into Indonesia. The Mount Toba Volcanic Eruption in Indonesia might have wiped out much of the animal and human life west of it and spared those to the east in Indonesia, but in 2013 the idea was refuted by research which found that the artifacts found in India came from much earlier pre-modern homins which migrated out of Africa, the Neanderthals and the Denisovans, and that even they survived the volcanic fall out. It is possible that they passed the virus to modern humans in Indonesia long after they brought it from Africa. Evidence for this hypothesis is supported by the facts that between 4% and 6% of the genome of Melanesians (Papua New Guinean and Bougainville Islanders) is derived from a Denisovan population. This DNA was most likely introduced during the early migration to Melanesia. These findings agree with the results of other tests which show a relative increase in genetic sharing between the Denisovan and the Aboriginal Australians when compared to other Eurasians and Africans. Nonetheless, the Papuans of Papua New Guinea have more Denisovan DNA than Aboriginal Australians. It appears pre-human or successor modern humans, infected with the virus and passing it vertically to their offspring, slowly crossed back to the Asia mainland and spread up the east coast of China to the Arctic and crossed the Bering Land Bridge from 45,000 to 12,500 years ago. The rattlesnakes, which are contemporary with modern humans, were probably infected by biting humans, eating rodents who obtained the virus from humans, or through human tick parasites they carried with them during the migration. Evidence for this retrograde movement of the paleovirus out of Indonesia back to the Asian mainland and India can be found in the snakes of the regions.”—The Black Dragon Trilogy,
https://a.co/a4UWw0x.
The five extinctions before were: Ordovician-silurian Extinction: 440 million years ago, small marine organisms died out; Devonian Extinction: 365 million years ago, many tropical marine species went extinct; Permian-triassic Extinction: 250 million years ago, largest mass extinction event in Earth’s history affected a range of species, including many vertebrates, by end of Permian, 251 million years ago, 96% of species lost, “the great dying”; Triassic-jurassic Extinction: 210 million years ago, extinction of other land vertebrate species allowed dinosaurs to flourish; Cretaceous-tertiary Extinction: 65 Million Years Ago, end of dinosaurs other than birds, allowing mammals to flourish.
Biodiversity and wild places provide the buffer and sources of control and balance that prevent and control emerging infectious disease. Three examples of how human influence yield biological toxic and infectious diseases, beyond the COVID-19 pandemic, are as follows. This summer the California Waterfowl Association, has reported the worst avian botulism outbreak in recent history at the Klamath Basin National Wildlife Refuge Complex. Thousands of acres of wetland and marsh, usually rich and verdant havens for millions of migrating waterfowl and shorebirds, have become a wasteland of dried-up mud ponds strewn with dead ducks, geese, and other water fowl, victims of avian botulism, caused by toxin-producing bacteria, a disease which tends to affect wetlands lacking flowing water which is cool, clean, and aerated. Thanks to drought and limited water, a direct result of extraordinary temperatures and climate change, these bad conditions are exactly what have developed this summer in the refuge. The only good news, working in the makeshift field hospitals of PVC-framed tents and shallow swimming pools for the birds, at least 90 percent of the collected living birds were rehabilitated and returned to the wild in botulism-free parts of the refuge. However, field surveyors have collected about 20,000 dead birds, and they guess at least twice as many may have perished.
Even good intentions of human intervention in wild places may amplify an infectious disease problem. At a meeting on Rickettsia and Rickettsial Diseases, Marseille, France, in May 2008, a paper was presented describing how the Brazilian government decided to kill off a large portion of the giant rodent Capybara population, a reservoir for spotted fever, which kills up to 30% of people infected if untreated. When they did this, ticks in the area carrying spotted fever bacteria actually increased. They found out, like all rodent populations, killing off adults increases reproduction of those that remain, decreasing immunity to spotted fever because of more young naive individuals, in turn, infecting more ticks with higher doses of bacteria.
Going back even further, yet not as long ago as one might expect, are human influences on emergence of epidemics of plague. “Although the recurrence and extent of plague epidemics in Europe, and eventually the Americas, continued to decline and become less extensive, China, the origin of plague (also, alternatively, to China, or elsewhere, from near Lake Issyk Kul in Kyrgyzstan in 1338–1339) continued to have large outbreaks of plague killing at least 10 million people between 1855 and 1959. How could this have happened in such contrast to Europe and elsewhere? A recent study in 2014 published in the Proceedings of the Royal Society B, based on very thorough historical records from China, which allowed the investigators to reconstruct the plague’s transmission routes across China during this period, indicates the “cause” was socioeconomic. They concluded that plague spread along established transportation routes, major roads, rivers, and coastlines, and moved with the greatest speed along these routes. Weather also played a role. Heavy rains and flooding increased the movement and infectious rate to and in new areas. It is reasonable to assume the floods forced people and their social parasites, rodents to transport the plague carrying fleas and infected individuals to new locales.” The Black Dragon Trilogy by JOHNATHAN KIEL
https://a.co/cxSzMtY.
Plague outbreaks are still a yearly occurrence in the Island of Madagascar due to its severe ongoing ecological damage. The plague is mostly endemic in Congo, Peru and Madagascar; the latter saw a large outbreak in 2017 with more than 2300 cases and 202 deaths, according to WHO. In 2017, Madagascar experienced two concurrent outbreaks of plague: a
flea-transmitted bubonic plague outbreak that spread beyond the usual endemic rural areas into new rural, as well as urban, areas; and a second highly-contagious human to human transmitted pneumonic plague outbreak which spread rapidly predominantly in three main urban areas of the country.

Human influence on the environment is like playing a child’s game of pick up sticks, pulling out the “wrong sticks” leads to collapse of a system, releasing a catastrophic chain of events, famine and even war, including epidemics and pandemics. Environmental quality decline, including climate change, and destruction of wild places, predominantly for agriculture to feed populations in sensitive areas, using poor and clear cutting methods expanding search for more farm land, following loss of arable land to drought and nutrient exhaustion, is leading to the sixth mass extinction, aided by the release of pandemic disease. As climate change increases drought and global warming, desertification increases, causing human and animal migrations, spreading disease, disease vectors and wild place destruction. We can aid this or learn to mitigate its progression or, if we fail, face another mass extinction which will replace us with a new dominant species.


Inactivation of Infectious Agents in the Environment is Exponential: The Initial Numbers Really Matter: Controlling Initial Contamination of SARS-CoV-2 is Critical
The decline in infectious agents from viruses to bacteria to toxins in the environment is not linear. The decline in SARS-CoV-2 from droplets to further traveling dehydrated small particles is not just based on physical deposition but also from ultraviolet light, other factors, and inherent instability. Regardless of the cause, in general, the decline commonly follows an exponential decay curve. Therefore, based on this model, social distancing does not lead to a zero dose of virus at six feet or any determinable distance. However, infectious dose or measurable recoverable virus (as by PCR) falls below a detection threshold.
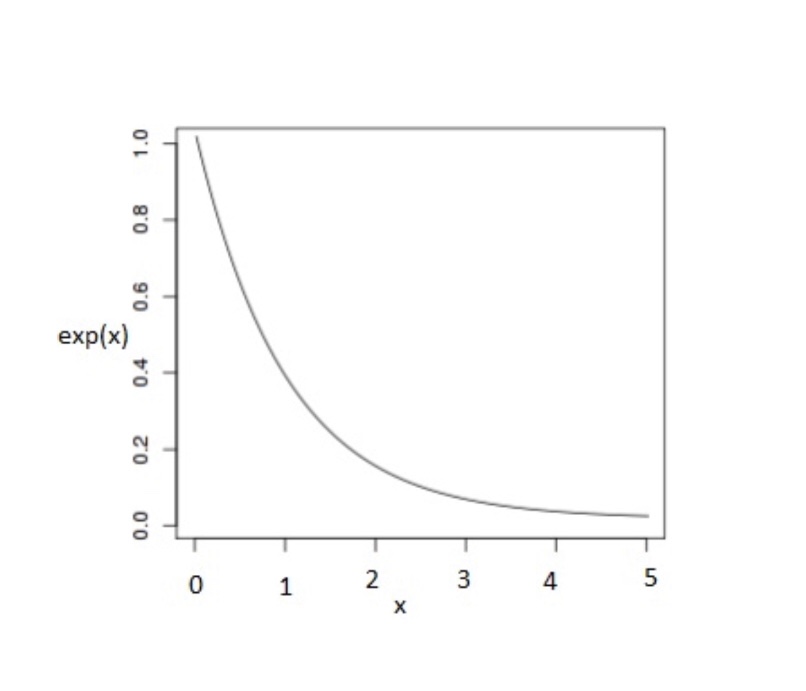
Is there real data to support this model? Actually, there is and for an infectious agent much more durable than SARS-CoV-2, anthrax spores:
The bottom line is that the viral load dispensed, even falling off exponentially, can extend for a longer time and distance above the infectious threshold as the initial amount of virus increases. This result means that the initial amount released is vitally important for determining subsequent exposures and number of people infected. By wearing masks and practicing good hygiene, the released dose of virus and its dwell time in the air and general environment can be dramatically decreased, with dramatically fewer infections. The opposite increases the time and distance of dangerous exposure.
Nanobes: A Solution to Fighting Viruses like SARS-CoV-2 and Other Pathogens Has Deep Roots in Basic Science
Nanobes, which were invented over many years research at Brooks AFB, San Antonio, Texas, (DALM, an essential component of Nanobes, first accidentally synthesized March 1, 1988) with roots before that at the Departments of Biochemistry and Microbiology at Texas Tech Health Sciences Center, Medical School, from 1977 to 1981, culminated with the co-invention of an extension of Chemically Programmable Immunity invented by the Nobel Laureate Dr Kary Mullis (American biochemist. who, in recognition of his invention of the polymerase chain reaction (PCR) technique, shared the 1993 Nobel Prize in Chemistry with Michael Smith) Both our experiences as chemists (mine with a PhD in biochemistry) made this approach a reality in biomedical applications. My veterinary medicine (DVM) and microbiological (PhD) training facilitated the medical applications. Let me first lay the foundation for the final product of our efforts, quantum theory. What is quantum mechanics or theory? The probability of finding a subatomic particle within a space or determining the velocity of the same particle of a known mass within the same space but not both at the same time, as defined by Johnathan L Kiel. This oversimplified description will suffice to introduce the basic science and subsequent basic research roots of the applied science. It was first described in my book Type B Cytochromes: Sensors and Switches (CRC Press, 1995) and then to completion in Nanowarfare, third book of The Black Dragon Trilogy (Amazon eBooks, 2018). The best description of this journey is summed up as follows:
“I learned that diffusion and Brownian motion, as described by Einstein, swept out volumes and brought about interactions some of which resulted in chemical reactions which were determined in part by the cross sections of the molecules and then by the respective kinetic energies they retained in a population. His description of these interactions actually proved molecules and atoms were in fact real entities and not just mathematical notions. For me, this approach showed that for particles larger than molecules and representing a phase interface, as between enzymes or reactive molecules immobilized on a solid surface interacting with other molecules in solution, surface area is a dominant factor in determining the kinetics of interaction. Therefore, a smaller and smaller particle, short of a molecule or atom, such as a nanoparticle, becomes more and more reactive based on the total surface area generated by the division into such particles. Second, my inspiration from his work on the photoelectric effect and the Quantum Theory to which it led, led me to understand the quantum effects seen in DALM, even to the point that a micro to nano particle could demonstrate its own unique quantum effects beyond those of the electrons of atoms and molecules. The DALM polymers, in nanoparticle form, even demonstrated Einstein’s quantization of heat capacity. The culmination of these two aspects, inspired by Einstein’s work, over the course of my scientific career and investigations, led me not to a dead end but to the total concept of Nanobes. They have become a bridge between classical and quantum physics with accompanying chemistry imposed on biology.”— The Black Dragon Trilogy by JOHNATHAN KIEL
https://a.co/i37MY9v
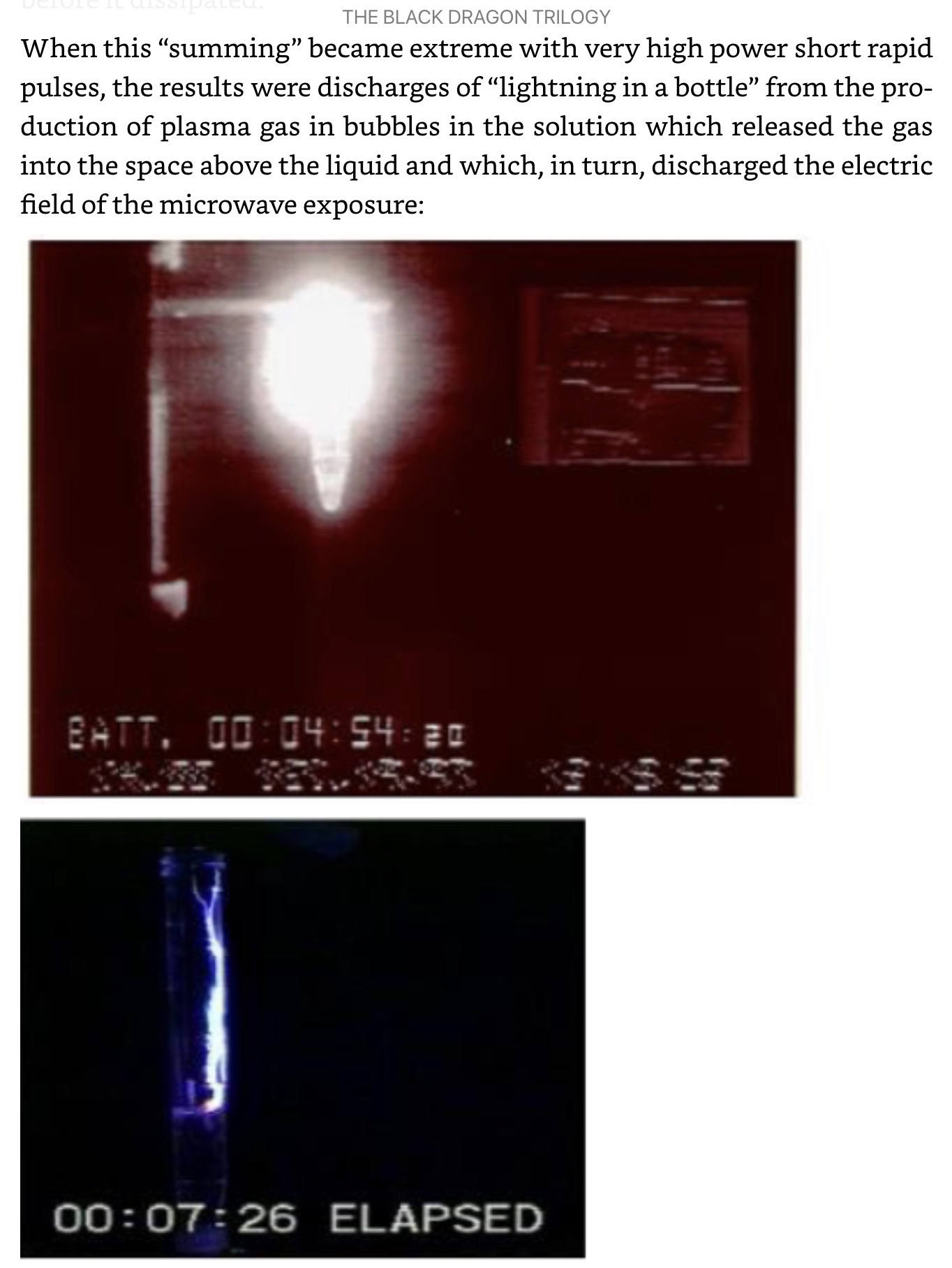
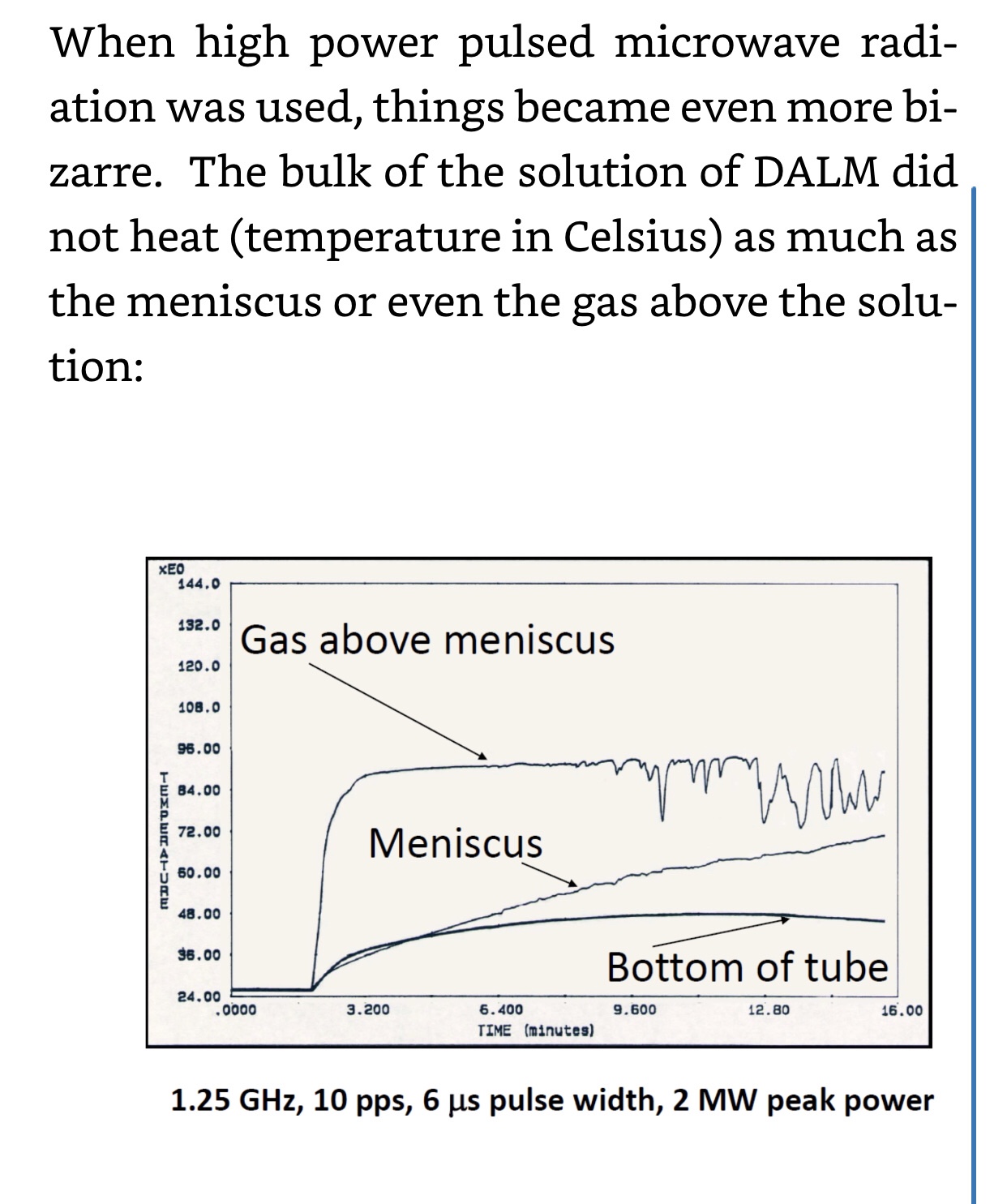
Demonstration of quantum effects in the polymer DALM which composes Nanobes: “how far apart the electron pair is spread by being trapped in or out of plane rings, they can interact and re-admit light at a later time as long as nothing affects their states in the meantime. This has led to the observation of the amazing ability of DALM to remember what it has been exposed to and to store energy. If that energy is stored and released suddenly, such as with a high energy microwave pulse, then an explosive reaction occurs in the DALM solution compressing gas bubbles of carbon dioxide, nitrogen and oxygen by the accompanying shock wave and producing plasma gas (ionized gas) which glows and even emerges to the surface of the solution where, in the presence of the strong electric field of the microwaves, is discharged in a spectacular demonstration of lightning in a bottle,”— The Black Dragon Trilogy by JOHNATHAN KIEL
https://a.co/5jZ4kha