No one can deny the great achievements of Pfizer and Moderna in developing vaccines that provide protective immunity to COVID that appear, based on the data they have presented, to be more than 90% and 94% effective, respectively. They both involve the delivery of mRNA which translate into in situ in vivo SARS-CoV-2 spike protein immunogens. Both vaccines because of the inherent instability of RNA need “cold chains” that are more demanding than for current vaccines for other viral diseases. Moderna’s vaccine remains stable in conventional refrigerators (4C) for a month and ordinary freezers (-20C) for six months. The Pfizer–BioNTech vaccine must be stored at -70C. The latter requires an ultra low freezer. This is because RNA is sensitive to environmentally abundant RNAase enzymes and oxidative damage, which break it down or renders it unusable. How can this be, if viruses like coronaviruses, contain RNA which does not suffer this fate as easily? There are even infectious agents that are composed entirely of RNA (viroids) which survive ordinary environmental conditions and the onslaught of host cell ribonucleases. In fact, many viruses produce their own ribonucleases to breakdown host cell messenger RNA (mRNA). Are there lessons to be learned from these agents that would lead to more stable vaccines, even to those that would not need a cold chain at all ? BioNTech/Pfizer and Moderna encapsulated their mRNA vaccines within lipid nanoparticles while the University of Oxford/Astrazeneca and CanSino incorporated antigen-encoding sequences within the DNA carried by adenovirus to yield mRNA to produce the spike protein immunogen. These physical barriers protect the mRNA until it has to be exposed while being translated into protein by host ribosomes, Novavax placed external recombinant S proteins of SARS-CoV-2 onto their proprietary virus like particle nanoparticles, providing direct stimulation to the immune system. All these to provide stability to the RNA and some direct immunogens, and consistency in dose delivery. Nanoparticles can codeliver adjuvants to help prime the desired immune responses, as well. Adjuvants are immunostimulatory molecules administered together with the vaccine to help boost immune responses mainly by activating additional molecular receptors that predominantly recognize pathogens or danger signals, but are inherently, in themselves, nonspecific. While the vaccine stimulates recognition and response of lymphocytes, in preference to innate cells, the activation of the innate immune cells is required to activate the lymphocytes, both B and T-cell responses. Encapsulation and/or conjugation of both the adjuvant and antigen or mRNA within the same nanoparticle enables targeted, synchronous delivery to the same antigen presenting cells (APC). Uptake of the antigen and adjuvant by APCs at separate times, can sometimes lead to autoimmunity against host proteins because the adjuvant can activate APCs that are not primed against the immunogen, but rather against self-antigens. BioNTech/Pfizer, composed their SARS-CoV-2 receptor binding domain (RBD) immunogens onto a T4 fibritin-derived “foldon” trimerization base to resemble the trimeric natural structure of the spike protein. Fibritin is a structural protein of bacteriophage T4, which catalyzes a phage-assembly process. It promotes the assembly of the long tail fibers of the bacteriophage and their attachment to the tail baseplate which attaches to host bacteria; it is also a sensing device that controls the retraction of the long tail fibers in adverse environments and prevents infection of target bacteria under those unfavorable conditions https://pubs.acs.org/doi/pdf/10.1021/acsnano.0c07197.
All this containment and, as mentioned in a previous post, certain sequence changes to discourage adverse innate or off target adverse effects, have only limited effect on stability. Viruses have developed several strategies to protect the stability of their RNA including inherent RNA shields, appropriating host RNA stability factors, incapacitating host RNA degradation and changing the general host RNA stability in cells using virally encoded nucleases. Viral RNA with poly(A) tails of the transcript, form an inhibitory structure for ribonucleases. Another common RNA structure that provides protection from a class of RNA degradation enzymes is the stable terminal stem loop found at the 3’ end of all viruses that lack a poly(A) tail. These structures prevent exosome-associated exoribonuclease from attacking viral RNAs, thereby keeping the 3’ end intact and blocking the 3’to-5’ degradation of the transcript.
Some viruses encode proteins that specifically protect viral RNAs from degradation during infection. An example of this is the nucleocapsid protein of negative sense RNA viruses such as vesicular stomatitis virus which assembles on the viral genome and antigenomic RNAs cotranscriptionally and renders the RNA resistant to RNases. Another example is the KSHV ORF57 protein, which binds to a specific viral element and protects intronless transcripts from degradation in the nucleus. In summary, these inherent defense mechanisms seem to be used by numerous viruses as a general way of protecting non-coding RNAs or RNAs, that are not translated, from degradation by the cellular RNA degradation machinery. This assists in the accumulation of viral templates for replication, transcription and packaging. Viral mRNAs, however, do not seem to employ this strategy for stabilization because it might be largely incompatible with efficient translation by cellular ribosomes. Instead, using host cell RNA stability factors is preferable for the latter. One attractive strategy for viruses to confound host cell RNAase enzymes is for their mRNAs to use cellular factors whose natural function is to stabilize cellular transcripts. The two most widely studied cellular RNA stability factors are HuR and poly(C)-binding protein 2 (PCBP2). The ubiquitously expressed HuR protein has been demonstrated to bind and stabilize well over 50 independent mRNAs by interacting with uridine-rich or adenine-uridine-rich elements. Alphaviruses such as Sindbis virus have been shown to contain high-affinity U-rich HuR protein-binding sites in their 3’ untranslated regions (UTRs) and to use HuR protein to stabilize viral mRNAs and promote a productive infection. Segmented RNA viruses remove the caps along with a small portion of the 5’ UTR from cellular transcripts and incorporate them into their own transcripts to facilitate the stability and translation of viral mRNAs. RNA viruses also encode their own ribonucleases or factors that can stimulate RNA degradation. Cap-stealing endonucleases encoded by many segmented RNA viruses, for example, selectively target cellular mRNAs for decay by 5’-to-3’ exonu- cleases and this dramatically changes the regulation of mRNA stability in infected cells. Segmented RNA viruses use the capped oligomers generated by endonuclease cleavage to initiate transcription of their own mRNAs. The nucleocapsid protein of Lassa fever virus, a member of the Arenaviridae, has recently been shown to possess 3’-to-5’ exonuclease activity. This exonuclease is essential for the virus to interfere with aspects of the interferon and other innate immune responses. The only other RNA virus known to date to encode an exonuclease is the Nsp14 protein of the SARS coronaviruses. This protein is similar to that of the Lassa fever virus nucleocapsid protein, thereby, also probably attacking RNAs from the 3’ end using a catalytic mechanism with two metal ions. Small RNA from viral genomic transcripts can also aid in protecting viral RNA. All insect-borne flaviviruses use the cellular Xrn1 5’-to-3’ exonuclease to generate a small flavivirus RNA (sfRNA) from the 3’ UTR of the virus. This small RNA is generated because of a set of pseudoknot structures capable of stopping the exonuclease. The small sfRNA generation is important for viral replication and cytopathology in West Nile Virus infection. Furthermore, instead of avoiding the RNAi (interference RNA) machinery, HCV, another member of the Flaviviridae, uses the abundant liver miRNA-122 to increase the accumulation of its mRNA during infection. miRNA-122 binds to two sites in the 5’ UTR of HCV and forms a unique structure required for efficient viral replication. Finally, viruses use nucleases to increase the rate of viral RNA evolution. Mutations in the Nsp14 exonuclease of independent coronaviruses result in a 15–21-fold decrease in replication fidelity. Therefore, the presence of this exonuclease can clearly influence viral RNA mutation rates and the generation of quasispecies https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3123725/pdf/main.pdf.
Viroids are small circular RNAs that mostly infect plants, mimic DNA in secondary structure in order to use host DNA-dependent RNA polymerase to replicate and do so by a rolling circle method that does not require the ring of RNA to open. The linear replicates are then ligated into new circles by a host ligase. This process avoids open ends that would be attacked by cell exonucleases and the necessity for start/stop sequences. They encode a hammerhead ribozyme which can cut and splice RNAs. The replication cycles of viroids include, for members of Pospiviroidae, replication in an asymmetric cycle. DNA-dependent RNA polymerase (polII) transcribes mature circular RNA into oligomeric (−) intermediates and these into oligomeric (+) intermediates. The (+) intermediates are enzymatically cleaved into monomers and ligated with DNA ligase 1 into mature circles. Members of Avsunviroidae replicate in a symmetric cycle where circular RNA is transcribed into oligomeric intermediates by nuclear-encoded polymerase (NEP). The intermediates are cleaved into monomeric units by the internal hammerhead ribozyme and ligated by host tRNA (transfer RNA) ligase into circles. The presence of thermodynamically stable as well as metastable structures of RNA are essential through the entire process so RNA structure determines fitness for replication. Virusoids are essentially viroids that have been encapsulated by a helper virus coat protein (providing a viral physical barrier to protect stabilize its RNA). They are thus similar to viroids in their means of replication (rolling circle replication) and due to the lack of expressible genes (into protein) but they differ in that viroids do not possess a protein coat https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6237808/.
Artificial stability can be built into synthetic RNAs. This has been necessary for diagnostic and especially therapeutic use of aptamers. These are small synthetic nucleic acids selected in vitro from a large library of sequences for their ability to bind to proteins and other targets specifically and strongly and then to be amplified by PCR. Derivatives or replacements of the phosphate and sugar backbone while maintaining base integrity allows them in many cases to still replicate. Some of these modifications are phosphorothioate (PS) bond substituting a sulfur atom for a non-bridging oxygen in the phosphate backbone of an oligonucleotide; and 2′-O-Methyl (2’OMe), a naturally occurring post-transcriptional modification of RNA, 2’OMe found in tRNA and other small RNAs. Oligonucleotides can be directly synthesized to contain 2’OMe. This modification increases the Tm (melting temperature) of RNA:RNA duplexes, but results in only small changes in RNA:DNA stability. It prevents attack by single-stranded endonucleases, but not exonuclease digestion. 2′-fluoro bases have a fluorine-modified ribose which increases binding affinity (Tm) and also confers some relative nuclease resistance compared to native RNA. Inverted dT can be incorporated at the 3′ end of an oligonucleotide, leading to a 3′-3′ linkage that will inhibit degradation by 3′ exonucleases and extension by DNA polymerases. In addition, placing an inverted, 2′,3′ dideoxy-dT base (5′ Inverted ddT) at the 5′ end of an oligonucleotide prevents spurious ligations and may protect against some types of enzymatic degradation. Phosphorylation of the 3′ end of oligonucleotides will inhibit degradation by some 3′-exonucleases. Phosphoramidite C3 Spacer can be incorporated internally, or at either end of an oligo to introduce a long hydrophilic spacer arm for the attachment of fluorophores or other pendent groups. The C3 spacer also can be used to inhibit degradation by 3′ exonucleases. https://www.idtdna.com/pages/education/decoded/article/modification-highlight-modifications-that-block-nuclease-degradation. Other modifications include the replacement of the phosposaccharide backbone with a polypeptide. For aptamers and Nanobes some of these modifications and others have been employed:
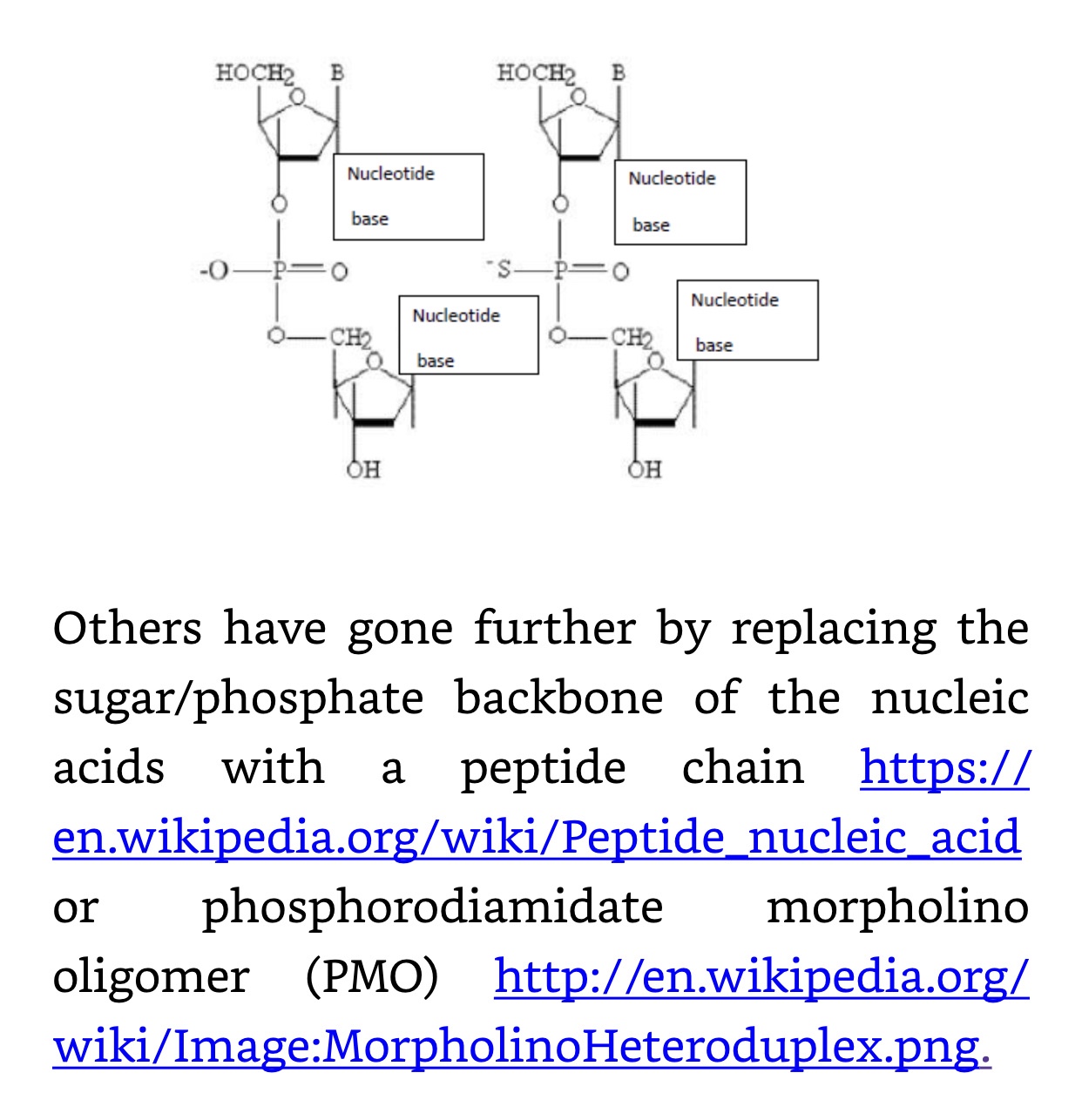
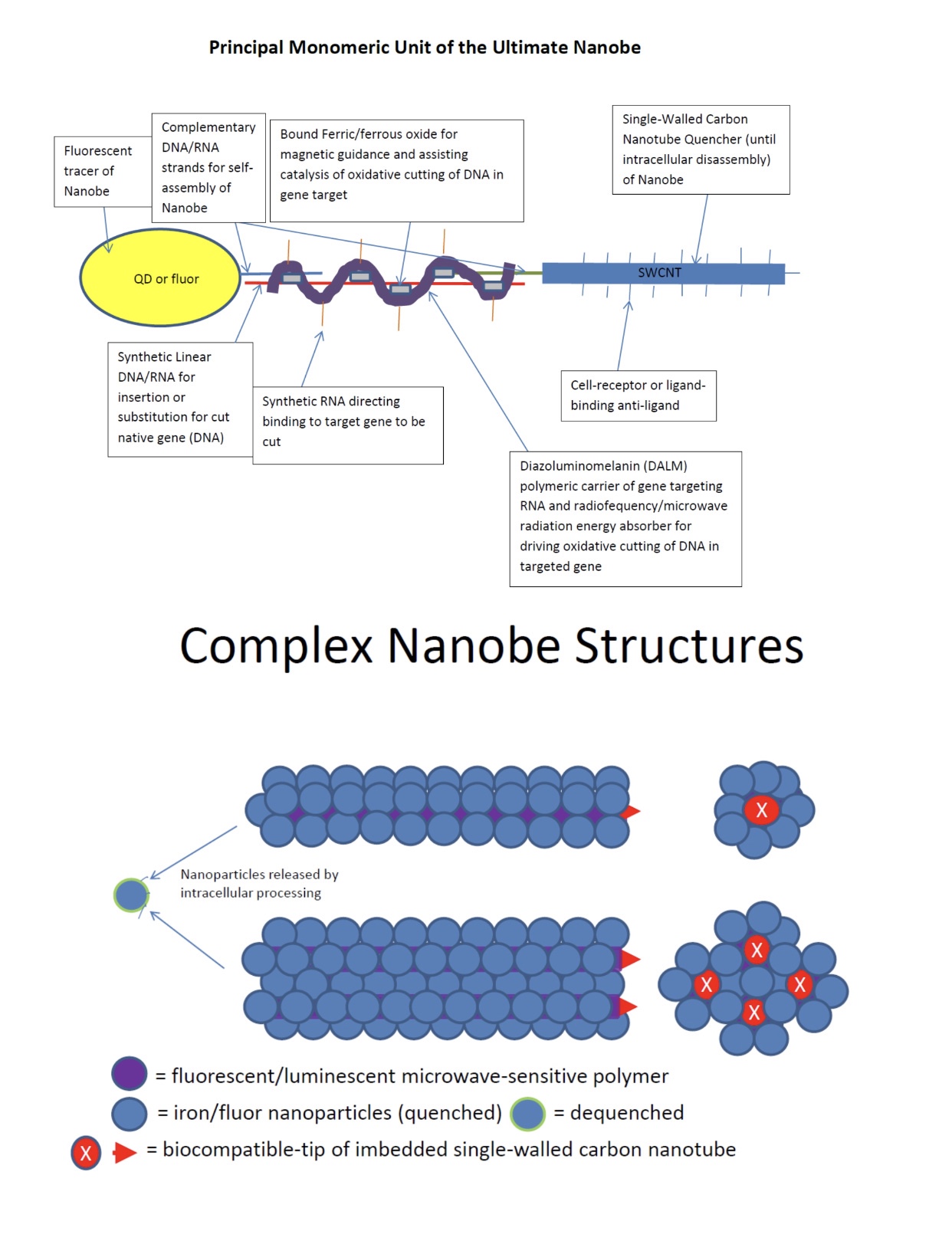
In Nanobes, we chose double-stranded DNA aptamers to provide the most stable unmodified synthetic nucleic acids. However, these are still vulnerable to nucleases of certain types. Also, DALM, in Nanobes, chelates metals that are necessary to certain nucleases, inhibiting them, but not all non-metal nucleases. The final modification in Nanobes that has yet to be tried is replacement of circular RNA or DNA with micro linear nucleic acid capped at the ends with carbon nanotubes and/or nanoparticles:
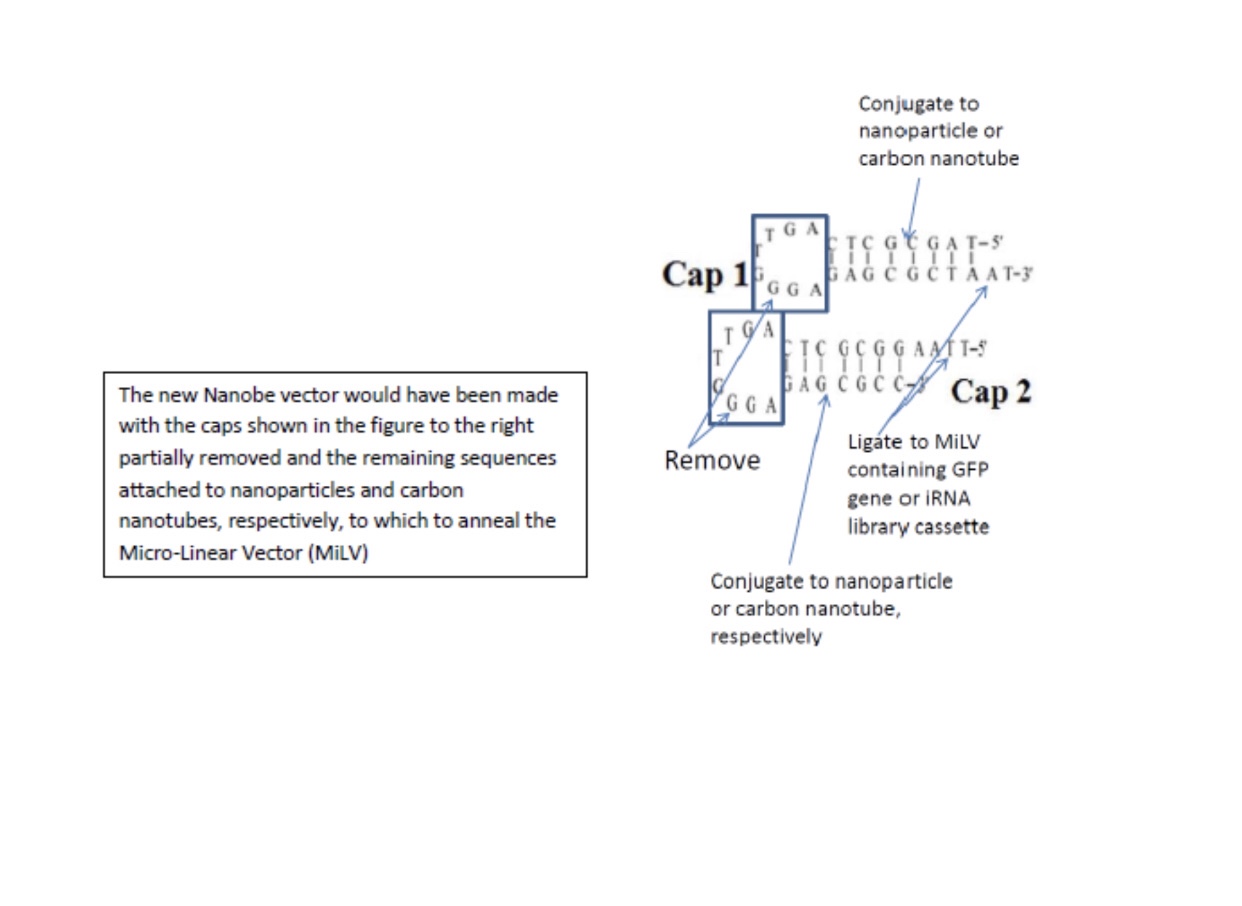
Nanobes in general do not require lipids to enter cells and can have aptamers on their surfaces which direct them to certain cells to maximize therapeutics or vaccines to optimal effects.